The Departments of Anesthesiology and Psychiatry at Washington University School of Medicine have established a multidisciplinary center that combines basic research, clinical research, clinical pain management and education in an organized, interactive group of dedicated researchers and clinicians.

Basic Research
Our basic research is concentrated in areas that are most likely to provide insights into fundamental aspects of chronic pain conditions, to complement activities in clinical research and practice.
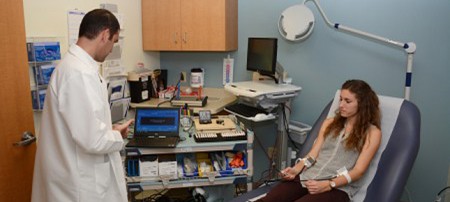
Clinical & Translational Research
Clinical research involves studies to better understand mechanisms of disease in humans; investigation and refinement of new tests and technologies in humans; testing of pharmacological and non-pharmacological interventions in individuals with or without disease; behavioral and observational studies; as well as outcomes research and health services research.

Center for Clinical Pharmacology
The Center for Clinical Pharmacology is advancing the rational use of medicine, translating basic research into actionable, clinical research and care, and preparing the next generation of clinical scientists.
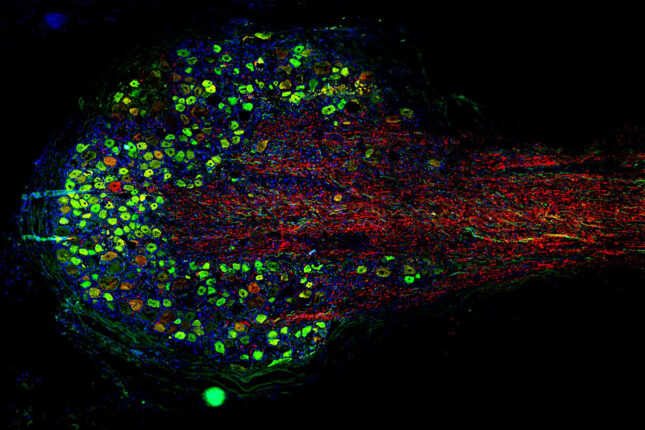
Integrated Research Center for Human Pain Tissues (INTERCEPT) Pain Center
The INTERCEPT Pain Center investigates alternative approaches to managing pain, particularly chronic pain. The center aims to develop innovative, evidence-based strategies that can effectively alleviate pain and reduce the need for opioids.
There are multiple positions open at all levels for scientists interested in joining our mission to understand mechanisms of pain and develop new treatments. Learn more >>